Research Article
Primer Pairs for Rice (Oryza sativa L.) Bisulfite Sequencing Studies

Mehmet Karaca1* and Ayse Gul Ince2
1Department of Field Crops, Akdeniz University, 07070 Antalya, Turkey
2Vocational School of Technical Sciences, Akdeniz University, 07070 Antalya, Turkey
*Address for Correspondence: Mehmet Karaca, Department of Field Crops, Akdeniz University, 07070 Antalya, Turkey, Email: mkaraca@akdeniz.edu.tr
Dates: Submitted: 22 October 2018; Approved: 05 November 2018; Published: 06 November 2018
How to cite this article: Karaca M, Ince AG. Primer Pairs for Rice (Oryza sativa L.) Bisulfite Sequencing Studies. J Plant Sci Phytopathol. 2018; 2: 091-098. DOI: 10.29328/journal.jpsp.1001024
Copyright License: © 2018 Karaca M, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Differential methylation; Epigenetics; Gene element; Promoter; Touch-down PCR

Abstract
Many agriculturally important properties such as heterosis, inbreeding depression, phenotypic plasticity, and resistance for biotic and abiotic stresses are thought to be affected with epigenetic components. New discoveries related with epigenetics are likely to have a major impact on strategies for crop improvement in rice breeding. However, assessing the contribution of epigenetics to heritable variation in plant species still poses major challenges. Methylation of cytosine in DNA is one of the most important epigenetic mechanisms in plants. DNA methylation not only plays significant roles in the regulation of gene activity, but also it is related with genomic integrity. Although most of next generation sequencing (NGS) technologies do not require the use of target specific primer pairs to identify and study DNA cytosine methylation, validation studies of NGS uses selective primer pairs. Bisulfite sequencing technique is a gold method for DNA cytosine methylation studies. However, bisulfite sequencing requires the development of bisulfite primer pairs to selectively study DNA sequences of interest. In this study 9 bisulfite specific primer pairs were identified and validated. These primer pairs successfully amplified bisulfite converted and unconverted genomic DNA extracted from radicle and plumule of rice (Oryza sativa L.) seedlings. Results of the present study clearly revealed the occurrence of CG, CHG and CHH (H stands for C. T or A nucleotides) contents in studied DNA sequence targets were different indicating potential role of DNA cytosine methylation in these genes. Primer pairs reported in this study could be used to detect DNA methylation which is one of the most important epigenetic mechanisms affecting the development, differentiation or the response to biotic and abiotic stress in rice (Oryza sativa L.).
Introduction
Paddy rice (Oryza sativa L.) is grown in more than one hundred countries in the world with a total harvested area of approximately 158 million hectares, producing more than 700 million tons, 470 million tons of which are milled rice. Nearly 640 million tons of paddy rice are grown in Asia, followed by Sub-Saharan Africa that produces about 19 million tons and Latin America that produces about 25 million tons. Currently China is the world’s largest rice producer, accounting for 35% of total world’s rice production with a planting area of 29 million hectares [1].
Turkey is not among the larger rice producing countries. The Marmara-Thrace region of Turkey is the largest rice-growing area and makes the greatest contribution to production, followed by the Black Sea region. Paddy rice (rice in the husk) production of Turkey is about 750000-850000 tons from approximately 95000-100000 hectares planting area [2]. In Turkey, average yield of paddy rice per hectare ranges from 6.5 tones (Baldo variety) to 10 tones (Luna variety) while average milling rate ranges from 55% (Baldo variety) to 68% (Luna variety). Edirne province in the Thrace region produces more than 50 percent of Turkey’s paddy rice production while Central Anatolia (“Çankırı” and “Çorum” regions) produces nearly a quarter of paddy rice production. Rice varieties such as Osmancık 97, Baldo, Cammeo, Galileo, Ronaldo, and Luare have popularity in Turkey because of their high yields and high milling rates along with their better cooking and ϐlavor characteristics. Rice in Turkey is irreplaceable for making pilaf, rolling stuffed, stuffed vegetables and several desserts such as rice pudding. However, rice production of the Turkey is not sufϐicient to counteract its consumption which is about 790000 tons annually [3].
In many parts of the world including Turkey low and high temperatures limit and damage rice growth during the different stages of plant development. Low temperatures during germination and at the seedling negatively affect panicle formation, flowering and pollination, resulting in yield losses. On the other hands, high temperatures in many parts of rice production areas in the world especially in the southeastern part of world cause spikelet sterility. Also drought or water shortage, salinity and alkalinity negatively affect the rice growth and development. In Turkey, blast disease (Pyricularia oryzae), brown leaf spot (Helminthosporium oryzae) and root rot (Fusarium moniliforme) are among the most important rice diseases [2,3].
Many agriculturally important properties such as heterosis, inbreeding depression, phenotypic plasticity, and biotic and abiotic stresses are thought to be affected and regulated with epigenetic components. New epigenetic discoveries are likely to have a major impact on strategies for crop improvement in plant breeding. Most abiotic and biotic growth limiting factors are known to be under epigenetic regulations [4]. Aberrant DNA methylation is known associated with loss of DNA homeostasis and genomic instability leading to development of human diseases such as cancer. Epigenetic variation due to the DNA cytosine methylation and histone levels in the genome results from enzyme-mediated chemical modifications. Methylation of cytosine in DNA is one of the most studied and important epigenetic modifications in plants. In plant genomes, DNA cytosine methylation occurs at the C5 position of cytosines in the three sequence contexts or islets, CG, CHG and CHH where H is A, T, or C. In plants, Domains Rearranged Methyltransferase 2 (DRM2) plays major roles in de novo cytosine methylation. There are specific enzymes for methylation and maintenance among the three sequence contexts CG, CHG and CHH. For instance, DNA methyltransferase 1 (MET1) and chromomethylase 3 (CMT3) catalyze methylation of CG and CHG while CHH methylation is maintained through de novo methylation by DRM2 [5,6]. It is well known that DNA methylation not only plays significant roles in the regulation of gene activity, but it is also related with maintenance of genomic integrity. Therefore, the studies of DNA cytosine methylation offer important finding for gene expression and regulation [7,8].
Currently there exist several techniques for assaying and detecting global and gene-specific cytosine DNA methylation in plant species. Among these techniques bisulfite sequencing is the gold standard for assessing DNA methylation. Bisulfite sequencing uses bisulfite-mediated deamination before sequencing. It could reveal the methylation status of every cytosine nucleotide of a genome under study. It was developed by Frommer and colleagues and recognized as a revolution in DNA methylation analysis. It is based on conversion of genomic DNA by using sodium bisulfite application [8]. This method takes the advantage of the selective and complete conversion of unmethylated cytosine to uracil by sodium bisulfite application. In this method converted cytosine nucleotides are amplified using polymerase chain reactions (PCR) as thymine nucleotides [7]. PCR amplified products are cloned (or directly sequenced) and, the sequencing experiments reveal the initial methylation profile of the region of interest [8,9].
Reliability and success of bisulfite-mediated deamination and sequencing method is strongly dependent on the availability of primer pairs that could uniquely and successfully amplify bisulfite treated and untreated genomic DNA region. By selecting specific primer pair any gen structure and regulatory sequences could be studied. The aim of this study was to develop a set of bisulfite specific primer pairs using DNA sequences of rice deposited in NCBI GenBank database and validate these primers on rice cultivar Osmancik 97.
Materials and Methods
DNA Sequences utilized
Genomic DNA sequences of 8 different rice genes (Oryza sativa L.) deposited in NCBI GenBank databases (https://www.ncbi.nih.gov/) were downloaded. Downloaded DNA sequences were used as input files for EpiOne software [8]. The occurrence of methylation context, CG, CHG and CHH were calculated and occurrences and densities were identified for each gene elements.
Sequences obtained from EpiOne were used to design primer pairs specific to bisulfite sequencing studies using Primer 3 software [10] based on the following main parameters: GC content value of a primer was set between 40% and 80%, annealing temperature (Tm) was set between 58°C and 62°C, and expected amplified product size was defined as 300–800 bp. After designing the sequences, cytosine bases (C) in the forward primers were replaced with Y (C/T) and the guanine bases (G) in the reverse primers were replaced with R (A/G). In the primer sequences R stands for purine (A or G) and Y stands for pyrimidine (C or T).
Genomic dna isolation and bisulfite treatment experiments
Genomic DNA samples of rice cultivar Osmancik 97 were extracted from plumule and radicle sections of seedlings using a protocol previously described in Karaca et al. [11]. Genomic DNA samples obtained from plumule and radicle sections were bisulfite treated using a bisulfite conversion kit (Invitrogen Corp., Carlsbad, CA, USA) as previously described in Karaca et al. [12].
A touch-down polymerase chain reaction (Td-PCR) was performed in a 25 μL reaction volume consisting of 3 µL bisulfite converted or control genomic DNA as the template, 0.5 μM forward and reverse primers, 80 mM Tris-HCl (pH 8.8), 19 mM (NH4)2SO4, 0.009% Tween-20 (w/v), 0.28 mM of each dNTP, 3 mM MgCl2, and 1 unit of Taq DNA polymerase (Invitrogen Corp., Carlsbad, CA, USA).
The Td-PCR amplification profile used in the present study was as follows: initial denaturation at 94°C for 3 minutes, ten cycles with denaturation at 94°C for 30 seconds, annealing at 60°C (or 66°C) for 30 seconds in the first cycle, diminishing by 0.5°C each cycle, and extension reactions at 72°C for 1 minute using a Veriti 96-well thermal cycler (Applied Biosystems, Foster City, CA, USA). Additional 40 more PCR cycles were run using the same cycling parameters mentioned above with constant annealing at 55°C (or 60°C). Denaturation and extension conditions were the same as indicated above. The amplification reactions ended with final extension reactions at 72°C for 10 minutes PCR products in the presence of 1x DNA loading buffer were loaded in 3% (w/v) high-resolution agarose gels (SERVA Electrophoresis GmbH, Heidelberg, Germany) and electrophoresed at 5 V per cm at constant voltage for 4-6 hours [8].
Electrophoretic purification, cloning, purification and sequencing experiments
PCR products of bisulfite treated and untreated targets (control or reference DNA) amplified with four different primer pairs were added with 5 μL 6x DNA loading buffer, mixed well and loaded in 3% (w/v) high-resolution agarose gels and electrophoresed at 5 V per cm of gel at constant voltage for 4-6 hours. PCR products in the gels were transferred into 2 mL tubes and purified using a PureLink Quick Gel Extraction & PCR Purification Combo Kit (Invitrogen Corp. Carlsbad, CA, USA). Purified amplicons were ligated into pTZ57R/T, vectors and were transformed to E. coli bacteria strain JM107 using a Transform Aid Bacterial Transformation Kit (Thermo Scientific, Waltham, MA USA). Targets carrying colonies were selected and sub-cultured and plasmid DNA samples were extracted using a GeneJET Plasmid Miniprep Kit (Thermo Scientific). A total of 12 plasmids with PCR fragments from bisulfite treated genomic DNA and 8 plasmids with PCR fragments from untreated DNA were sequenced using M13R sequencing primers (Macrogen Inc., Amsterdam, The Netherland) [12].
Sequences of plasmids were downloaded from Macrogen Inc. and primer sequences of forward and reverse were identified from each clone sequence and trimmed off along with the vector sequences. Target sequences were formatted into fasta file using Sequencher program and analyzed using the KisMeth software program [13]. The methylation status of each target was calculated for each cytosine sequence context (CG, CHG and CHH) using the percentage methylation (%), calculated as 100×C/(C+T). The statistical significance between and within the samples of the entire set of either CG, CHG or CHH sites was separately evaluated with the nonparametric Mann-Whitney U test. In this study, two-tailed p values, 0.05 were considered statistically significant. Methylation context (CG, CHG or CHH) was used as factor and the methylation percentage was use as response.
Results and Discussion
In this study, a total of seven genes and regulatory sequences with different functions were studied. These genes were further mined into gen bodies and regulatory sequences using EpiOne software program [8]. Based on the EpiOne programs, sequences were mined into gene structures or elements such as exons, introns, promoters and untranslated regions (UTRs). Also three Coding DNA Sequences (CDS) that were portions of mRNA transcripts translated by ribosomes were also studied (Table 1). Introns and exons are usually called as gene bodies that do not contain regulatory regions such as promoters and UTRs. Studies of DNA methylation in gene bodies provide further advantages [14] since type, amount, and location of DNA cytosine methylation within a gene provides significant knowledge on the enzymatic pathway.
| Table 1: Sources of 18 primer pairs, gene, gene bodies, promoters and related information. |
|
||||||||
| GI | Primer ID | Gene | Gene Body | Range (bp) | Target Range (bp) | Tm (oC) |
Target Size (bp) |
|
|
| 1449931 | OS-F-001 OS-R-001 |
Sucrose phosphate synthase gene | Intron | 2001/2638 | 2052 2621 |
59.99 59.98 |
570 |
|
|
| OS-F-002 OS-R-002 |
Exon | 3173-3862 | 2933 3675 |
59.88 60.08 |
743 |
|
|||
| OS-F-005 OS-R-005 |
Exon | 5236/6177 | 5593 6253 |
60.97 60.03 |
661 |
|
|||
| OS-F-006 OS-R-006 |
Exon | 6499/7150 | 6432 7147 |
59.52 61.11 |
716 |
|
|||
| 4165327 | OS-F-007 OS-R-007 |
3-hydroxy-3-methylglutaryl-CoA reductase gene | Exon | 2898/3910 | 2658 3387 |
60.04 59.99 |
730 |
|
|
| OS-F-008 OS-R-008 |
Intron | 2898/3910 | 3352 4063 |
59.97 59.97 |
712 |
|
|||
| OS-F-009 OS-R-009 |
Exon | 4258/5123 | 4035 4766 |
59.86 59.94 |
732 |
|
|||
| OS-F-010 OS-R-010 |
Intron | 4258/5123 | 4955 5596 |
60.09 61.17 |
642 |
|
|||
| 21727819 | OS-F-011 OS-R-011 |
Partial BP-73 gene for SAP-like protein | Intron | 2045/4515 | 1912 2582 |
60.52 60.97 |
671 |
|
|
| OS-F-012 OS-R-012 |
Intron | 2045/4515 | 2554 3152 |
60.99 59.96 |
599 |
|
|||
| OS-F-013 OS-R-013 |
Exon | 4516/5621 | 4458 5093 |
58.04 58.05 |
636 |
|
|||
| OS-F-014 OS-R-014 |
Exon | 4516/5621 | 5060 5758 |
59.75 60.32 |
699 |
|
|||
| 25264667 | OS-F-015 OS-R-015 |
Partial tub A2 gene for tubulin alpha | Intron | 3323/4214 | 3094 3879 |
59.77 59.99 |
786 | ||
| OS-F-016 OS-R-016 |
Intron | 3323/4214 | 3820 4287 |
60.09 60.24 |
468 | ||||
| OS-F-017 OS-R-017 |
Exon | 4215/4493 | 3786 4478 |
59.89 60.55 |
693 | ||||
| 56410257 | OS-F-018 OS-R-018 |
CDPK2 gene, promoter region and 5’- UTR | CDS | 1/1080 | 144 807 |
59.97 59.92 |
664 | ||
| 115253055 | OS-F-019 OS-R-019 |
Partial tub6 gene for beta-tubulin 6 exon 1-2 | CDS | 1/2836 | 2207 2773 |
60.45 59.96 |
567 | ||
| 657189313 | OS-F-020 OS-R-020 |
Granule-bound starch synthase (Wx) gene, 5’-UTR | CDS | 1/2694 | 1438 2035 |
59.52 59.95 |
598 | ||
| Please note that Tm: melting temperature. GI: gene identification number of GenBank, CDS: Coding DNA Sequence. Target size is the size of amplified product (bp), UTR: untranslated region. | |||||||||
Occurrence of GC, GHG and GHH islets among sucrose phosphate synthase, 3-hydroxy-3-methylglutaryl-CoA reductase, BP-73 gene for SAP-like protein, tub A2 gene for tubulin alpha, promoter region and 5’- UTR of CDPK2, tub6 gene for beta-tubulin 6 exon 1-2 and granule-bound starch synthase (Wx) with 5’-UTR were different as calculated with EpiOne program. This indicated that occurrence and densities of these methylation specific islets within gene bodies and regulatory regions could play significant roles in regulation of these genes studied in rice gene expression [12-14].
We could design more primer for some genes while some had fewer number of primers (Table 1). We strictly avoid CG dinucleotides, potential hairpin structures and possible primer dimers. Amplification of targets used touch-down PCR conditions. This help us not to rely on a gradient PCR thermocycler to determine the appropriate annealing temperature. Primer pairs from intron, exons and regulatory sites were designed so that DNA methylation could be targeted at specific regions on a gene element (either exon, intron or UTRs). In the present study, we also used a strategy similar to that of primer walking to cover whole intronic or exonic regions for a gene under study. Gene targets used in this study are among genes that are important for rice yield and productivity. For instance, sucrose synthase gene is very important for sucrose which is the major photosynthetic product in many plant species. Also sucrose is main sugar for translocation of carbohydrate form leaves to tissues such as grain in rice. Furthermore, sucrose is also known a signal molecule that regulates gene expression in plants [15]. Therefore, information regarding the epigenetic and genetic regulation of sucrose synthase gene could be used in the studies of rice improvement plant protection and productivity.
Target genes also included 3-hydroxy-3-methylglutaryl-CoA reductase (rice HMGR like) and granule-bound starch synthase (Wx) gene. The gene HMGR like coded in rice has been found to increase expression not by wounding but pathogen, Magnaporthe grisea, a well-known causal agent of rice blast disease [16]. This suggested that this enzyme in rice might play a key role as a component of the inducible defense mechanism, therefore, epigenetic studies of this gene may provide information in understanding of epigenetic regulation. In rice as well as in other cereals, granule-bound starch synthase gene is responsible for amylose synthesis. This enzyme is regulated at the transcriptional and post-transcriptional levels but its epigenetic regulation is not known. Since this enzyme affects the ratio of amylose to amylopectin in grain. Understanding the regulation of granule-bound starch synthase activity may lead to the improvement of rice eating quality [17].
A total of 18 bisulfite specific primer pairs were initially designed and they were first tested using rice genomic DNA sample that was not bisulfite treated (called control or reference DNA). Genomic DNA samples were amplified using two different touch-down PCR profiles. Primer pairs; OS-FR-11 and OS-FR-13 both targeting intronic and exonic region in BP-73 gene for SAP-like protein were failed to amplify targets in both touch-down amplification profiles (Ann 60 and Ann 66). On the other hand, OS-FR-14 which was also failed to amplify target, could amplify target when annealing temperature reduced to 50oC. Primer pairs OS-FR-15, OS-FR-19 produced an extra amplicon at Ann 60, but extra amplicons were vanished away when these primer pairs were used at Ann 66 amplification profile.
Among 16 primer pairs, which were found to successfully amplified genomic DNA samples of variety Osmancik 97, those OS-FR-01, OS-FR-02, OS-FR-07, OS-FR-09, OS-FR-10, OS-FR-12, OS-FR-16, OS-FR-18 and OS-FR-20 could also amplify bisulfite treated genomic DNA samples. These primer pairs were called bisulfite specific primer pairs and listed in Table 2. On the other hands, 7 primer pairs failed amplification of bisulfite treated genomic DNA probably due to the original C richness creates long stretches of uracil, which might result in DNA fragmentation during the bisulfite-treatment. We assumed that fragmentation of targets were encountered in those targets greater lengths. This is why bisulfite sequencing studies use short segments of genomic region in order to avoid failed amplification due to the bisulfite treatments. During the bisulfite treatment DNA fragmentation occurs and this may reduce or remove the number of available molecules for PCR amplification [12].
| Table 2: Bisulfite specific primer pairs for rice. | ||||
Primer ID |
Forward (F) and Reverse (R) primer sequences |
Tm (oC) | TD Profile | Size |
OS-F-001 |
GTTGATTTAATTTAGTTTYTYGTGYTY |
59.99 | Ann 60 | 570 |
RCAATCTCARAAACAAATACCTTTTAR |
59.98 | |||
OS-F-002 |
TGYAAAATTTTAYTGAGTTYYTTTATT |
59.88 | Ann 60 | 743 |
RATCTTRTAARTTRARTCAATCTCCTC |
60.08 | |||
OS-F-007 |
YAAATYTAAGGTTGGATGGTAAGATAA |
60.04 | Ann 60 | 730 |
AAAAACCACACCATTAATAARAACAAR |
59.99 | |||
OS-F-009 |
GTYATTAGTAGAATTGAAYGTYATYAA |
59.86 | Ann 60 | 732 |
AACATRTACTARCTAAACACATCATRC |
59.94 | |||
OS-F-010 |
YGTGTGTTTTATTAATGGYTTAATYTT |
60.09 | Ann 60 | 642 |
TAAATATRRCCACTCAACTCTACTRAR |
61.17 | |||
OS-F-012 |
TAYAYTGTTTATGAGTGTGYYATTTTA |
60.99 | Ann 60 | 599 |
CTATRRAAAARAACATATCTARCCTTR |
59.96 | |||
OS-F-016 |
TYGTTGYAGAGATTATTAGGAGTTAAT |
60.09 | Ann 60 | 468 |
CAGTCTCACTRAARAARRTRTTAAAA |
60.24 | |||
OS-F-018 |
TYGTTGYAGAGATTATTAGGAGTTAAT |
59.97 | Ann 60 | 664 |
AATAAACTCAACACACAACATCARAAR |
59.92 | |||
OS-F-020 |
CCTTYAGTTYTTTGTYTATYTYAAGA |
59.52 | Ann 60 | 598 |
TRAATTTCAATCTTARCTTTRACCTAT |
59.95 | |||
| Please note that within the degenerate primer sequences, R stands for purine (A or G) and Y stands for pyrimidine (C or T) and H stands for A, T or C. TD: touch-down PCR profile, Tm: melting temperature. Ann stands for annealing temperature at 60 oC. | ||||
In order to validate bisulfite specific primer pairs developed in this study, primer pairs OS-FR-02 and OS-FR-10 were used. OS-FR-02 amplified exonic region of sucrose phosphate synthase gene while OS-FR-10 amplified intronic region of 3-hydroxy-3-methylglutaryl-CoA reductase gene (Figure 1). As shown in Figure 1, DNA cytosine methylation in the intronic region of 3-hydroxy-3-methylglutaryl-CoA reductase gene was not statistically different between radicle and plumule of rice variety Osmancik 97. On the other hands, methylation of CHH were different between radicle and plumule of rice variety Osmancik 97. Methylation of CG and CHG were not statistically differed between radicle and plumule of rice variety Osmancik 97. Based on the high level of occurrence and densities of methylation islets in 7 gene regions that we did not validate in the present study we expected that their use in DNA methylation studies in rice would reveal valuable information. Also primer pairs reported in this study could be used as molecular markers in genetic and breeding studies [18].
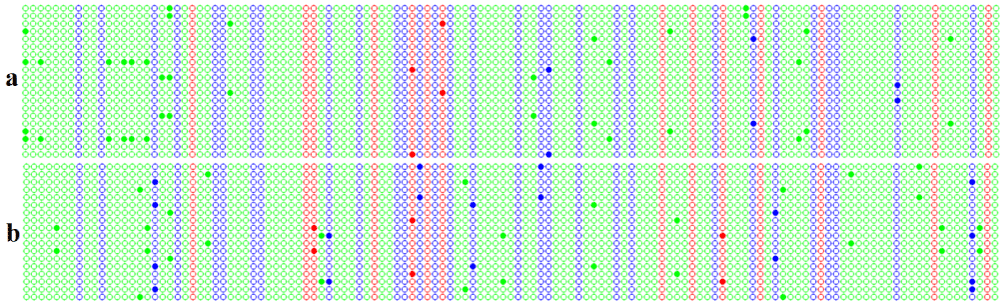
Figure 1: Methylation status of 3-hydroxy-3-methylglutaryl-CoA reductase gene between radicle and plumule tissues of rice cultivar Osmancik 97. Each circle in the panels represents a cytosine residue, methylated cytosine residues are shown with full circles, and non-methylated cytosine residues are shown with empty circles. Cytosine residues are color-coded by their sequence context: red for CG, blue for CHG and green for CHH where H is C, T or A. Each row of panels represents a sequence of clone.
In the present study selected genes are important genes for rice yield and yield related parameters. Previous research identified a number of epigenetic regulators in rice [19]. For instance, it was found that CG and CHG hypomethylation associated with activation of genes preferentially expressed in rice seed development. Other epigenetic regulators in rice include involve in a wide range of developmental and stress responsive pathways. Epigenetic variations detected with the use of primer pair reported in the present study could be used to identify epialleles related to important agronomic traits and may provide new strategies to enhance agronomically favorable traits and grain productivity in rice [15,19].
Conclusion
Increased yield and biotic and abiotic resistance in rice is one of the most desired research goals. Yield of rice can be determined using data of panicle number per plant, the filled grain number per panicle, and the weight per grain. Genetic and epigenetic regulation in these grain related characteristics are important for increased rice yield. Results revealed that methylation islets in the contexts of CG, CHG and CHH among 7 genes or regulatory regions consisting of sucrose phosphate synthase, 3-hydroxy-3-methylglutaryl-CoA reductase, BP-73 gene for SAP-like protein, tub A2 gene for tubulin alpha, promoter region and 5’- UTR of CDPK2, tub6 gene for beta-tubulin 6 exon 1-2 and granule-bound starch synthase (Wx) with 5’-UTR were different. We reported 9 gene specific primer pairs suitable in epigenetic studies in Osmancik 97 which is one of the most sown rice cultivars in Turkey. The use of these primer pairs could provide understanding of epigenetic regulation in rice, with an emphasis on the roles of key epigenetic regulators, the epigenomic landscape, epigenetic variation, transposon repression, and plant development. These primer pairs reported in the present study could be also used in the validation studies in high-throughput sequencing of rice genomes.
Acknowledgement
This work was partially supported by the Scientific and Technological Research Council of Turkey (TUBITAK, 113O935).
References
- Guo L, Liu M, Zhang Y, Tao Y, Zhang F, et al. Yield differences get large with ascendant altitude between traditional paddy and water-saving ground cover rice production system. European Journal of Agronomy. 2018; 92: 9–16. Ref.: https://goo.gl/T9AVoZ
- Anonymous, USDA Foreign Agricultural Service, Global Agricultural Information Network Report. 2017; No: TR7014. Ref.: https://goo.gl/44h7Dc
- Anonymous, FAOSTAT Food Agricultural Organization. 2018; UN. Ref.: http://faostat.fao.org/
- Deng X, Song X, Wei L, Liu C, Cao W. Epigenetic regulation and epigenomic landscape in rice. National Science Review. 2016; 3: 309–327. Ref.: https://goo.gl/cwtgTq
- Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature Reviews Genetics. 2010; 11: 204-220. Ref.: https://goo.gl/73Le67
- Shen H, He H, Li J, Chen W, Wang X, et al. Genome-wide analysis of DNA methylation and gene expression changes in two Arabidopsis ecotypes and their reciprocal hybrids. Plant Cell. 2012; 24: 875-892. Ref.: https://goo.gl/WQyeY6
- Peng H, Zhang J. Plant genomic DNA methylation in response to stresses: potential applications and challenges in plant breeding. Progress in Natural Science. 2009; 19: 1037-1045. Ref.: https://goo.gl/Cs1Y5Z
- Karaca M, Ince AG. EpiOne: A software tool for identification of potential cytosine DNA methylation marks in promoters and gene bodies. Journal of Scientific and Engineering Research. 2016; 3: 295-301. Ref.: https://goo.gl/Jpt7Nx
- Jin X, Pang Y, Jia F, Xiao G, Li Q, Zhu Y. A potential role for CHH DNA methylation in cotton fiber growth patterns. PLoS ONE. 2013; 8: e60547. Ref.: https://goo.gl/AAwGBH
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods in Molecular Biology. 200; 132: 365-386. Ref.: https://goo.gl/fSisb9
- Karaca M, Ince AG, Elmasulu SY, Onus AN, Turgut K. Coisolation of genomic and organelle DNAs from 15 genera and 31 species of plants. Anal Biochem. 2005; 343: 353-355. Ref.: https://goo.gl/NGZn1Y
- Karaca M, Ince AG, Uygur-Gocer E, Aydin A. Bisulfite primer pairs for analysis of cotton (Gossypium spp.) DNA methylation. Journal of Scientific and Engineering Research. 2016; 3: 656-662. Ref.: https://goo.gl/VPJcV6
- Gruntman E, Qi Y, Slotkin RK, Roeder T, Martienssen RA, Sachidanandam R. Kismeth: analyzer of plant methylation states through bisulfite sequencing. BMC Bioinformatics. 2008; 9: 371. Ref.: https://goo.gl/9qVRHL
- Bewick, AJ, Schmitz RJ. Gene body DNA methylation in plants. Current Opinion in Plant Biology. 2017; 36: 103-110. Ref.: https://goo.gl/Yfb22X
- Yonekura M, Aoki N, Hirose T, Onai K, Ishiura M, et al. The promoter activities of sucrose phosphate synthase genes in rice, OsSPS1 and OsSPS11, are controlled by light and circadian clock, but not by sucrose. Frontiers in Plant Science. 2013; 4:31. Ref.: https://goo.gl/gcEN6h
- Nelson AJ, Doerner PW, Zhu Q, Lamb C. Isolation of a monocot 3-hydroxy-3-methylglutaryl coenzyme A reductase gene that is elicitor-inducible. Plant Molecular Biology. 1994; 25: 401-412. Ref.: https://goo.gl/AppLJJ
- Liu DR, Huang WX, Cai XL. Oligomerization of rice granule-bound starch synthase I modulates its activity regulation. Plant Science. 2013; 210: 141-150. Ref.: https://goo.gl/wo9Vad
- Ince AG, Karaca M, Uygur Gocer E, Aydin A. Descriptive statistics and PIC values of genomic-and transcriptomic-microsatellites in several plant species. Journal of Scientific and Engineering Research. 2017; 4:236-246. Ref.: https://goo.gl/cLGgeS
- Chen X, Zhou DX. Rice epigenomics and epigenetics: challenges and opportunities. Curr Opin Plant Biol. 2013; 16:164-169. Ref.: https://goo.gl/Y9qAqr